Rolandická epilepsie (dětská benigní parciální epilepsie s centro-temporálními hroty, benigní parciální epilepsie Childhoos s centro-temporálními hroty, BECTS)
Rolandická epilepsie (benigní parciální epilepsie Childhoos s centrotemporálními hroty, BECTS) je nejčastějším syndromem dětské idiopatické parciální epilepsie a jedním z nejsnáze rozpoznatelných epileptických syndromů. Toto onemocnění postihuje 15–25 % dětí školního věku s epilepsií (Heijbel et al., 1975; Dalla Bernardina et al., 2005).
Předpokládá se, že syndrom je geneticky podmíněný, ale mechanismus dědičnosti, dominantní nebo multifaktoriální, není jasný; to druhé se zdá pravděpodobnější. Výsledky nedávné studie dvojčat (Vladlamundi et al., 2006) vyvolaly pochybnosti o genetické povaze a spekulace o možném významu získaných faktorů. Problém je komplikován tím, že u méně než 10 % dětí s rolandickými hroty se rozvinou záchvaty a EEG rysy pravděpodobně nejsou dědičné. Ložiska charakteristických rolandických ostrých vln jsou často detekována u dětí bez epilepsie, a proto je diagnostická hodnota při izolaci v izolaci.
Onemocnění začíná téměř vždy ve věku od 2 do 13 let, vzácné případy se vyskytují přibližně v jednom roce věku. Typické jsou „prosté“ parciální záchvaty, tedy probíhající bez poruchy vědomí. Jsou charakterizovány především motorickými projevy, i když častá je parestézie tváří a rtů a převážně se jedná o orofaryngeální svaly, což vede k slinění a/nebo zástavě řeči. Jednotlivé záchvaty jsou krátkodobé (30-60 sekund). 60 až 80 % záchvatů se vyskytuje během spánku nebo po probuzení.
Sekundární generalizace se rozvíjí u 20 % pacientů většinou uprostřed noci, zatímco motorické záchvaty se častěji objevují při usínání nebo probouzení. Může se objevit postiktální přechodná paréza (Toddova obrna). U většiny dětí se záchvaty vyskytují vzácně, dobře reagují na antiepileptika a během léčby je nízká epileptogenicita ohniska, které zůstává klinicky „tiché“ ve více než 90 % případů (Arzimanoglou et al., 2004; Dalla Bernardina et al., 2005).
Neurologické vyšetření pacientů s rolandickou epilepsií neodhalí žádnou patologii; Ve většině případů neurologický vývoj také probíhá normálně. Bylo však pozorováno mírné až středně těžké poškození řeči; Několik prací pojednává o kognitivní výkonnosti v BECTS (Staden a kol. 1988; Deonna 2000; Saint-Martin a kol. 2001a; Pinton a kol. 2006). V kontrolované studii Weglage et al. (1997) často nacházeli vazomotorické poruchy a problémy s prostorovým vnímáním u dětí s centrotemporálními vrcholy.
Povinnou změnou na EEG je přítomnost specifického typu fokálních epileptiformních výbojů: fokální bifázické pomalé vrcholy, střední až vysoké napětí, následované pomalými vlnami lokalizovanými v centrotemporálních oblastech, případně s difúzí do přilehlých oblastí, objevující se na pozadí normálního činnost na pozadí. U mladších pacientů dochází k zadnímu posunu v lokalizaci. Tyto výboje se mohou vyskytovat izolovaně nebo v krátkých sériích. Často je pozorováno dočasné vymizení a migrace paroxysmů z jedné hemisféry na druhou. Rozložení pole je jako horizontální dipól (pozitivita ve frontální zóně, maximální negativita v rolandické oblasti).
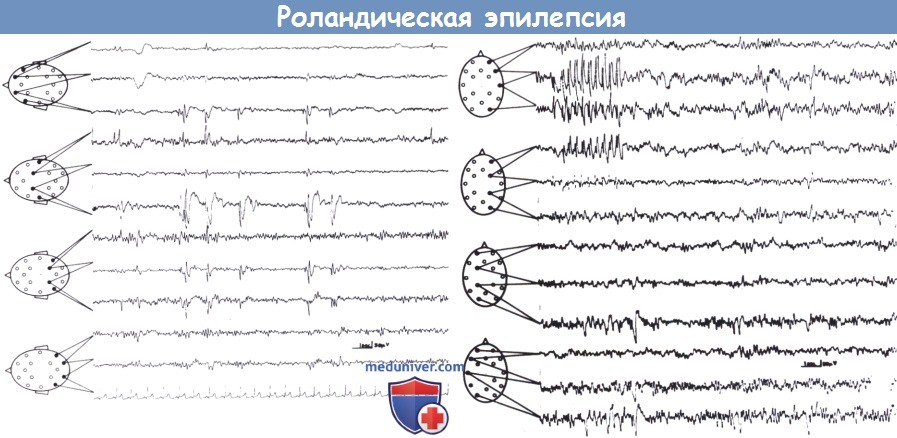
Benigní parciální epilepsie s centrotemporálními hroty.
Zaostřeno na levé straně u osmiletého chlapce (nahoře).
Pravostranné ohnisko se sérií opakujících se rolandických adhezí a vln v pravé centrotemporální oblasti u XNUMXletého chlapce (dole).
V obou případech paroxysmální aktivita jednoznačně odpovídá dolním rolandickým, midtemporálním elektrodám.
Neustálá aktivace během spánku. Lokalizace v horní rolandické oblasti je také možná, ale střední oblast se netýká (Legarda et al., 1994). Neobvyklá umístění však nejsou neobvyklá (Wirrel et al., 1995) a tvar vrcholů je pro diagnostiku důležitější než jejich přesná topografie. Přesto jsou paroxysmy, zcela identické s těmi, které se vyskytují u benigní parciální epilepsie, zřídka detekovány u epilepsií způsobených mozkovými lézemi (Gobbi a kol., 1989; Santanelli a kol., 1989; Ambrosetto, 1992). Jak uvádí několik autorů (přehled viz Dalla Bernardina et al., 2005), nejvýraznějším projevem centrotemporálních vrcholů je významné zvýšení jejich frekvence během ospalosti a ve všech fázích spánku. Přibližně u 30 % dětí se vyskytují pouze během spánku.
Výsledek Rolandické epilepsie s ohledem na záchvaty je mimořádně příznivý. Až 25 % dětí má pouze jeden záchvat, většina má více záchvatů; Ačkoli se záchvaty u některých pacientů vyskytují často, nemá to vliv na výsledek onemocnění. Relapsy po 16 letech jsou extrémně vzácné (Loiseau et al., 1988). Po léčbě rolandické epilepsie byly u několika dospělých pacientů pozorovány izolované generalizované tonicko-klonické záchvaty a bylo také hlášeno několik případů status epilepticus. Navzdory výše uvedeným potenciálním problémům je prognóza školního učení a sociálního fungování dobrá (Loiseau et al., 1983).
Několik výzkumníků hlásilo atypické projevy benigní parciální epilepsie (Aicardi a Chevrie, 1982; Fejerman a kol., 2000; Saint-Martin a kol., 2001a). U několika dětí s rolandickými ložisky a řídkými fokálními nočními záchvaty se mohou vyvinout atonické a/nebo myoklonické záchvaty, které se často vyskytují v sériích a vyskytují se mnohokrát denně; tento stav byl nazýván atypickou benigní parciální epilepsií (Aicardi, 2000; Fejerman a kol., 2000) nebo pseudo-Lennoxovým syndromem (Hahn a kol., 2001) kvůli atonickým záchvatům a výrazným abnormalitám EEG.
Atonické záchvaty jsou zvláště výrazné a mohou se vyskytovat desítkykrát denně, často doprovázené pády. Série záchvatů může trvat 2-3 týdny v intervalech několika měsíců. U těchto dětí je EEG zaznamenané během spánku velmi podobné EEG pacientů s prodlouženými vrcholovými vlnami během pomalého spánku (Aicardi a Chervie, 1982; Aicardi a Levy Gomes, 1992) a EEG při probuzení ukazuje mnohočetné bilaterální výboje komplexů hrotů -vlna. Kvůli pádům a silné záchvatovité aktivitě během spánku jsou takoví pacienti často mylně diagnostikováni jako Lennox-Gastautův syndrom. Přestože průběh tohoto syndromu může být benigní, se spontánní remisí před 10. rokem věku a po dvou nebo více sériích záchvatů, byly hlášeny případy s těžším výsledkem (Hahn et al., 2001). Kognitivní a behaviorální poruchy mohou přetrvávat, ale nebyly podrobně studovány.
Přesná nosologická definice tohoto syndromu dosud nebyla stanovena (Deonna et al., 1986), jednoznačně však patří do spektra epilepsií s kontinuálními spike-waves during sleep (CSWS). Částečný nebo sekundární generalizovaný myoklonus se může projevit jako atonické epizody. Takové epizody byly hlášeny u pacientů s benigní parciální epilepsií léčených karbamazepinem (Caraballo et al., 1989). Mnoho pacientů se syndromem však tento lék nedostalo (Aicardi a Levy Gomes, 1992).
Byly hlášeny epizody opercular status epilepticus zahrnující obličej, jazyk a faryngeo-laryngeální struktury (Saint-Martin et al, 1999). Byly doprovázeny rytmickými výboji hrotových vln, hroty byly synchronní s obličejovými křečemi. Během takových epizod mohou být pozorovány pseudobulbární symptomy, jako je slintání a dysartrie (Roulet et al., 1989; Boulloche et al., 1990; Deonna et al., 1993; Fejerman et al., 2000) a někdy se objeví skutečná afázie ( Roulet a kol., 1989).al., 1987). Kontrola některých z těchto epizod vyžadovala použití steroidů (Fejerman a Di Blasi, XNUMX).
Scheffer a kol. (1995b) pozorovali závažnou permanentní dyspraxii řeči a poruchy orální a lícní hybnosti u několika členů stejné rodiny s autosomálně dominantním mechanismem dědičnosti a možné anticipace; Guerrini a kol. (1999) uvádějí případ paroxysmální dystonie ve formě spisovatelské křeče a ataxie jako autozomálně recesivního syndromu, ale tento stav je odlišný od obvyklého benigního typu. Nedávno Roll a spol. (2006) identifikovali gen Xq22 SRPX2 jako způsobující rolandické záchvaty spojené s orální a řečovou dyspraxií a mentální retardací.
benigní rolandická epilepsie – geneticky podmíněná fokální epilepsie spojená s věkem podmíněnou hyperexcitabilitou centrálně-temporálního kortexu mozku. Benigní rolandická epilepsie se projevuje vzácnými, zejména v noci, křečovými záchvaty v polovině obličeje, jazyka a hltanu; v některých případech – generalizované epileptické záchvaty. Diagnóza se stanoví na základě klinických charakteristik onemocnění a údajů EEG, v případě potřeby se provádí polysomnografie a MRI mozku. Benigní Rolandická epilepsie nevede k poruchám v psychofyzickém vývoji dítěte a do konce dospívání mizí beze stopy.
ICD-10
G40.0 Lokalizovaná (fokální) (parciální) idiopatická epilepsie a epileptické syndromy se záchvaty s fokálním začátkem

Přehled
V pediatrii je benigní rolandická epilepsie nejčastější patologií ze skupiny benigních fokálních epilepsií. Tvoří 15 % všech případů epilepsie u dětí do 15 let a asi 20 % případů konvulzivního syndromu u dětí. Benigní rolandická epilepsie se vyskytuje s frekvencí 21 případů na 100 tisíc dětí. Benigní rolandická epilepsie se může objevit ve věku od 2 do 14 let, ale v 85 % případů se projeví mezi 4. a 10. rokem. Benigní průběh klasické Rolandické epilepsie je charakterizován absencí psycho-neurologických změn a úplným vymizením epileptických záchvatů do 15-18 let.
Příčiny
Přesné příčiny benigní rolandické epilepsie nebyly dosud stanoveny. V některých případech (podle různých zdrojů 20–60 %) lze vysledovat dědičnou povahu onemocnění, ale není pozorována přímá autozomálně recesivní nebo autozomálně dominantní dědičnost. To posloužilo jako základ pro polygenní teorii dědičnosti benigní rolandické epilepsie, podle níž je onemocnění určováno dvěma různými geny.
V moderní neurologii a epileptologii převládá představa, že benigní rolandická epilepsie vzniká v důsledku narušeného vyzrávání mozkové kůry v centrální temporální oblasti. Benigní rolandická epilepsie je onemocnění závislé na věku a je pravděpodobně spojeno se zvýšenou dráždivostí mozku u dětí. To je způsobeno řadou faktorů: funkčními a strukturálními rysy epileptogenních oblastí (neokortex, hippocampus, amygdala), prevalencí excitačních neurotransmiterů, nízkým obsahem GABA, nezralostí GABA receptorů a zvýšeným počtem excitačních reentrantních synapsí. Jak mozek dítěte dozrává, jeho dráždivost klesá a epileptogenita ložisek benigní rolandické epilepsie postupně mizí. V důsledku toho dochází k poklesu počtu epileptických záchvatů a úplnému uzdravení pacientů, když dosáhnou puberty.
Příznaky Rolandické epilepsie
Pro klasickou benigní rolandickou epilepsii jsou nejtypičtější jednoduché parciální epileptické záchvaty, které probíhají bez ztráty vědomí. Typicky záchvatu předchází smyslová aura ve formě jednostranného brnění, brnění nebo necitlivosti na obličeji, rtech, dásních, jazyku a krku. Dále dochází k motorickému paroxysmu, který může být reprezentován jak tonickými a klonickými svalovými kontrakcemi, tak i jejich kombinací. V závislosti na lokalizaci záchvatů mohou být záchvaty benigní rolandické epilepsie hemifaciální a faryngeálně-orální. Hemifaciální záchvat nastává s křečemi ve svalech poloviny obličeje. Faryngeální záchvat je charakterizován jednostrannými křečemi ve svalech jazyka, rtů, hltanu a hrtanu. Je doprovázena hypersalivací a poruchou řeči. Křečovité stahy svalů hrtanu vedou ke vzniku charakteristických hrdelních zvuků připomínajících klokotání, kloktání nebo chrochtání. Jak se rozvíjí benigní rolandická epilepsie, mohou záchvaty „změnit“ strany.
80 % epileptických záchvatů, které doprovázejí benigní rolandickou epilepsii, se vyskytuje v noci a úzce souvisí s obdobím usínání a probouzení. Mnohem méně častá je kombinace nočních záchvatů se záchvaty vyskytujícími se během dne. Ve 20 % případů se vyskytuje benigní rolandická epilepsie s brachiofaciálními záchvaty, při kterých se křeče obličejové oblasti šíří do homolaterálního ramene. V 8 % případů je pozorováno postižení homolaterální dolní končetiny. V některých případech je benigní rolandická epilepsie doprovázena komplexními parciálními epileptickými záchvaty, které se vyznačují krátkodobou ztrátou vědomí. Přibližně 20 % pacientů prodělá sekundární generalizované epileptické záchvaty, při kterých křeče zasahují všechny svaly těla. K takovým útokům dochází s úplnou ztrátou vědomí.
Aktuální
Benigní rolandická epilepsie se vyznačuje krátkou dobou trvání epileptických záchvatů (do 2-3 minut) a jejich nízkou frekvencí. Útoky mohou být sporadické nebo se mohou objevit několikrát do roka. Někdy na začátku onemocnění mohou být záchvaty častější, ale s přibývajícím věkem dítěte jsou méně časté.
Na rozdíl od Jacksonem popsané epilepsie není klasická benigní Rolandická epilepsie doprovázena opožděním psychomotorického vývoje dítěte a nevede k mentální retardaci. Nemůžeme však říci, že by došlo k absolutní absenci neurologických poruch, protože přibližně u 3 % pacientů dochází po záchvatech k přechodné hemiparéze.
diagnostika
Hlavní metodou diagnostiky benigní rolandické epilepsie je EEG. Protože u některých pacientů jsou změny EEG pozorovány pouze během spánku, je při normálním denním EEG navíc nutné noční EEG video monitorování nebo polysomnografie.
Patognomickým znakem benigní rolandické epilepsie je detekce ostrých vln s vysokou amplitudou nebo vrcholů lokalizovaných v centrálních časových svodech na pozadí normální základní aktivity. Po vrcholu často následují pomalé vlny, které spolu s vrcholem tvoří tzv. „Rolandský komplex“ trvající až 30 ms. Vizuálně takové komplexy připomínají komplexy QRST na elektrokardiogramu. Typicky jsou „rolandské komplexy“ lokalizovány na opačné straně, než jsou záchvaty, ale mohou být také bilaterální. Mezi rysy EEG vzorů benigní rolandické epilepsie patří jejich variabilita od jednoho EEG záznamu k druhému.
V první řadě je třeba odlišit benigní rolandickou epilepsii od symptomatických epilepsií, které se vyskytují u intracerebrálních nádorů, traumatických poranění mozku, zánětlivých lézí mozku (absces, encefalitida, hnisavá meningitida). Rolandická epilepsie je potvrzena nepřítomností patologických změn neurologického stavu a poruch chování, intaktní inteligencí, anamnestickými údaji a normální základní aktivitou na EEG. V některých případech se pro objasnění diagnózy provádí MRI mozku.
Diferenciální diagnostika benigní rolandické epilepsie a noční epilepsie s komplexními parciálními záchvaty představuje velké obtíže. Je to dáno tím, že poruchy řeči typické pro záchvat benigní rolandické epilepsie lze interpretovat jako poruchy vědomí. Na druhou stranu je poměrně obtížné adekvátně diagnostikovat noční poruchy vědomí. V takových případech hrají při stanovení diagnózy rozhodující roli EEG data, která u epilepsie frontálního a temporálního laloku prokáže ložiskové změny mozkové aktivity v odpovídajících svodech.
Určité obtíže způsobuje odlišení klasické benigní Rolandické epilepsie od pseudolennoxova syndromu, který je pozorován u 5 % pacientů s příznaky Rolandické epilepsie. Ve prospěch pseudolennoxova syndromu svědčí kombinace typických rolandických epileptických záchvatů s atypickými absencemi, myoklonickými a astenickými záchvaty, intelektuálně-mnestickými poruchami, ale i detekcí na EEG difuzní vrcholové aktivity či pomalých komplexů charakteristických pro Lennox-Gastautův syndrom.
Léčba benigní rolandické epilepsie
Pro dětské neurology a epileptology je otázka vhodnosti léčby benigní rolandické epilepsie značně kontroverzní. Vzhledem k tomu, že benigní rolandická epilepsie končí uzdravením i bez léčby, není podle některých autorů indikací k antiepileptické léčbě. Jiní neurologové s poukazem na pravděpodobnost diagnostického omylu při diagnostice „benigní rolandické epilepsie“ a s přihlédnutím k možnosti jejího přechodu do pseudolennoxova syndromu doporučují při opakovaných epileptických záchvatech antiepileptickou terapii.
V léčbě dětí s benigní rolandickou epilepsií se používá vždy pouze 1 antiepileptikum (monoterapie). Léčba obvykle začíná jedním z léků kyselinou valproovou. Pokud je to netolerovatelné nebo neúčinné, obvykle přecházejí na topiramát nebo levetiracetam. U dětí starších 7 let lze použít karbamazepin, ale je třeba si uvědomit, že v některých případech může vést k fenoménu zhoršení, tj. ke zvýšení frekvence záchvatů.





